
Isomer Generations using RDKit library for mass molecular docking tests (part-1)


Table of contents
I wrote a Python program to generate stereoisomers of compounds using the RDKit library and open-source docking software AutoDock Vina.
My primary focus was on Pantoprazole, a medication used to treat GERD (Gastroesophageal Reflux Disease), and I utilized its isomers to conduct molecular docking experiments on a proton pump inhibitor obtained from the PDB database
"The Python program generates stereoisomers when you input a compound string in the SMILES (Simplified Molecular Input Line Entry System) format."
My idea is to generate multiple isomers of a compound and then test for their molecular docking(Ligand-Receptor)
ex: Ex: Think of pantoprazole as a key and proton pump as a lock, so we are generating multiple keys based on the design of the previous key and trying a sort of hit-and-trial method to see if any of the newly generated keys with different structures will fit out lock.
From a theoretical perspective, it should work, as there is at least some probability of it working. However, it is challenging in practice because pantoprazole is specifically designed to fit the proton pump, and isomers have different shapes and configurations, so the probability of an isomer inhibiting the proton pump by binding to it is low.
Still, I wanted to try this to learn about the workings of RdKit, and It was fun to play around.
I learned so much during this endeavor of mine.
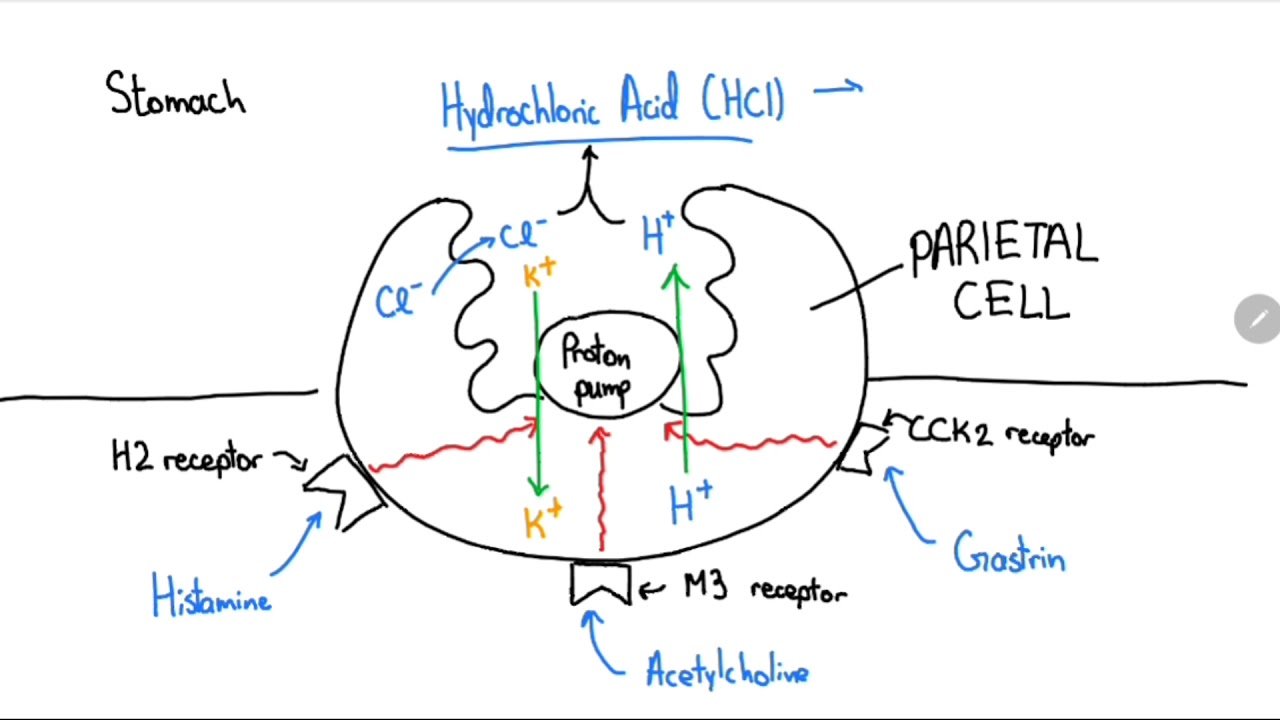
proton pump :
Code In Python.
import json
from rdkit import Chem
from rdkit.Chem import AllChem
from rdkit.Chem import Draw
# Defining the input molecule in SMILES format
# 2,3-dichlorobutane "CCC(Cl)C(Cl)C"
# pantoprazole -> "O=C(O)C(CC1=CC=C(C=C1)N)N[C@@H](C(=O)N)C(=O)N"
input_smiles = "COc1ccnc(c1OC)CS(=O)c2[nH]c3ccc(cc3n2)OC(F)F"
# Parsing the input SMILES to create an RDKit molecule
input_molecule = Chem.MolFromSmiles(input_smiles)
# Generate isomers
isomers = list(AllChem.EnumerateStereoisomers(input_molecule))
# dictionary to store isomers and their IUPAC-like names
isomer_data = {}
# Generate IUPAC-kinda names for each isomer and store them in the dictionary
for idx, isomer in enumerate(isomers):
iupac_like_name = Chem.MolToSmiles(isomer, isomericSmiles=True)
isomer_data[f"Isomer_{idx + 1}"] = {
"SMILES": Chem.MolToSmiles(isomer),
"IUPAC_Like_Name": iupac_like_name,
}
# Serialize the dictionary to JSON
json_data = json.dumps(isomer_data, indent=2)
# this Saves the json data to a file
with open("isomer_data.json", "w") as json_file:
json_file.write(json_data)
# visualize the isomers with diagram
for idx, isomer in enumerate(isomers):
img = Draw.MolToImage(isomer, size=(300, 300))
img.save(f"Isomer_{idx + 1}.png")
in this code, not only we are generating isomers, we are also saving them in an external JSON file and generating their images also, yaay.
The input will be a SMILES String, search about it to know more about what a SMILES format it
Thread:https://threadreaderapp.com/thread/1698701553727873100.html
Twitter link:
My Results :


This is Code Part
I will write 2 more blogs on this, explaining the theoretical workings of the RdKit Library and how it generates isomers and going through the internal workings of EnumerateStereoisomers function with an explanation of its GitHub code